香港氢能科学研究院「香港科大hkust研发全球最耐用氢燃料电池较铂催化剂高出37倍」
氢燃料电池利用氢及氧来发电,发电过程不会产生二氧化碳、悬浮粒子及其他有机会引致烟雾及健康问题的空气污染物,所以一直被视为较洁净的电能来源。虽然氢能对环境造成较少损害,亦已发展了一段时间,但始终未能大规模商业化。原因是氢燃料电池依赖电催化剂(electrocatalyst)发电。但催化剂一般由铂制成,这种金属不仅成本高昂,产量亦稀少。

香港科技大学(科大)的研究团队研发了一款崭新的氢燃料电池,不仅刷新了这种电池的最高耐久性记录1,且更具成本效益,有助推动绿色能源的普及化及实现碳中和目标。
氢燃料电池利用氢及氧来发电,发电过程不会产生二氧化碳、悬浮粒子及其他有机会引致烟雾及健康问题的空气污染物,所以一直被视为较洁净的电能来源。虽然氢能对环境造成较少损害,亦已发展了一段时间,但始终未能大规模商业化。原因是氢燃料电池依赖电催化剂(electrocatalyst)发电。但催化剂一般由铂制成,这种金属不仅成本高昂,产量亦稀少。
科学家们一直努力寻找譬如铁-氮-碳等较常见而廉价的物质作为铂替代品,惟这些物质的催化发电效能兼耐久性均欠佳。
近日,由科大化学及生物工程学系邵敏华教授带领的团队,研发出一个新配方,不仅大幅降低铂于催化剂的所需含量达八成,更刷新氢燃料电池发电耐久性的世界记录。
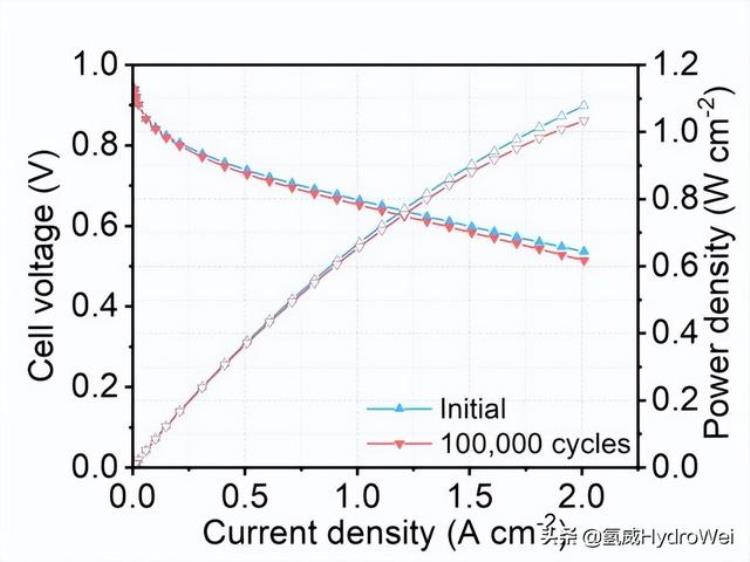
虽然这款新型混合催化剂的铂含量极低,但经过十万次电压循环的加速压力测试后,其催化效率仍维持在百分之97;而现时一般的催化剂经过了三万次的加速压力测试后,效率大跌逾五成。团队的另一项测试亦显示,氢燃料电池使用了新型混合催化剂后,即使持续运作超过200小时,催化效果也没有出现下降。
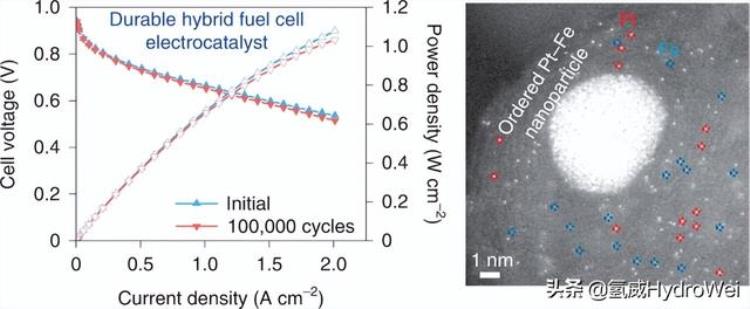
催化剂效能出众,其中一个原因是它有三个不同类型的活性中心(active site) 进行催化作用,较只有一个活性中心的传统催化剂多。由原子分散的铂、单原子铁,以及铂铁合金纳米粒子所组成的新型混合催化剂,可以加快催化速度,产生的催化作用亦较铂催化剂高出3.7倍。理论上,催化性能愈好,燃料电池所产生的功率也会愈大。
身兼科大能源研究院院长的邵教授表示﹕「要建立一个碳中和的社会,采用氢燃料电池这种绿色能源转换设备是有必要的。面对严峻的气候危机,绿色能源的应用必需进一步加强。我很高兴这个研究成果有助我们向这个目标迈进一步。感谢政府低碳绿色科研基金的支持,我们将继续优化催化剂,令它可应用于氢燃料电池车及其他电化学能源产品。」
研究获科技部国家重点研发计划、深圳市科技创新委员会及香港研究资助局拨出经费支持,有关研究内容近日刊登于期刊《Nature Catalysis》。
1根据美国能源部就氢燃料电池持久性的测试标准2把电压值设于0.6V保持三秒,其后再把电压值加大至0.9V再保持三秒为一次循环3电压值设于0.6V
传媒查询:林淑媛 电话﹕2358 6313电邮﹕anitalam@ust.hk 王敬莲电话﹕2358 6306/ 5190 7882电邮﹕lindywong@ust.hk
原子分散的Pt和Fe位点以及Pt-Fe纳米颗粒用于持久的质子交换膜燃料电池抽象质子交换膜燃料电池将氢气和氧气转化为电能,无排放。用于氧还原反应的Pt基电催化剂成本高、耐久性低,阻碍了其广泛的应用,非贵金属电催化剂的发展受限于其性能低下。在这里,我们设计了一种由原子分散的Pt和Fe单原子以及Pt-Fe合金纳米颗粒组成的混合电催化剂。其Pt质量活性是燃料电池中商业Pt / C的3.7倍。更重要的是,阴极中Pt负载较低的燃料电池(0.015mg铂厘米−2) 表现出出色的耐久性,在 100,000 次循环后具有 97% 的活性保持率,并且在 200 多个小时内在 0.6 V 时没有明显的电流下降。这些结果强调了混合电催化剂中活性位点间协同效应的重要性,并为为电化学器件设计更活性和更耐用的低Pt电催化剂提供了另一种方法。
 主要
主要
质子交换膜燃料电池(PEMFCs)作为一种有前途的清洁能源转换技术,已获得相当大的关注。然而,用于阴极氧还原反应(ORR)的Pt基纳米催化剂的高成本和低耐久性阻碍了该技术的广泛采用。1,2.根据30千瓦的最终成本目标−1用于燃料电池堆3,催化剂层中的Pt负载量必须低于0.125 mg cm−2(参考资料)4).然而,随着Pt负载的降低,由于可接近的活性位点有限,氧转移阻力增加,这导致耐久性降低4.因此,开发低Pt负载阴极的雄心在Pt利用和Pt基电催化剂的固有耐久性方面提出了巨大挑战。
尽管在开发先进的铂基催化剂方面付出了巨大努力,以提高铂的利用率和质量活性(MA)对ORR的影响5,6,在液体电池中测量的高活性和/或耐久性在燃料电池中很少实现。然而,由氮配位碳表面(Me-N-C)中高度分散的过渡金属单原子组成的碳基Pt基团无金属ORR电催化剂是取代Pt的有希望的候选者(ref.7).不幸的是,Me-N-C的耐久性差限制了它们的实际应用。8.一些早期研究9,10应用Me-N-C作为Pt基电催化剂的载体,旨在提高后者的稳定性。最近,Liu和同事们报告了一种具有超低Pt负载量(2-3重量%)的杂化催化剂,该催化剂由Co-N-C上负载的Pt-Co合金纳米颗粒组成,具有出色的ORR活性(1.77 A mg)铂−10.9 V 时无 iR,无压力校正)11.这一结果表明,即使少量的Pt引入也有助于提高混合电催化剂的高活性。尽管这种杂化 ORR 催化剂具有出色的 Pt MA,但在潜在循环(在 0.6 至 0.95 V 的 30,000 次循环后为 83%)和潜在保持(在 0.75 V 下 22 小时后为 45%),它仍然遭受了显著的活性损失(参考文献)。11).饶恩和同事12发现通过添加少量Pt(1-2 wt%)可以提高Fe-N-C的稳定性,尽管活性没有改变。
在这里,我们报道了一种混合电催化剂(表示为Pt-Fe-N-C),它由氮掺杂碳载体中高度分散的Pt和Fe单原子上的Pt-Fe合金纳米颗粒组成。多种类型的活性位点不仅使Pt MA高出3.7倍,而且具有出色的耐久性。即使在100,000个电位循环之后,性能损失也可以忽略不计,并且在具有超低Pt负载(0.015 mg)的燃料电池测试中,在0.6 V时没有观察到电流下降铂厘米−2) 在阴极中。
结果铂铁-N-C的结构和组成图1a显示了合成的Pt-Fe-N-C催化剂的典型透射电子显微镜(TEM)图像,该图像清楚地显示了主要尺寸分布为2-3nm的纳米颗粒(补充图1),分散在碳基底上,其Brunauer-Emmett-Teller表面积为750 m2g−1和介孔(补充图2)。Pt-Fe-N-C在相对较低放大倍率下的高角度环形暗场扫描透射电子显微镜(HAADF-STEM)图像(图1b和补充图3)清楚地表明除了纳米颗粒外,高密度的孤立原子锚定在碳基板上。金属间PtFe的峰集在Pt-Fe-N-C的X射线衍射图中得到了很好的分配(补充图4)。42.5°时的额外峰值可归因于无序的PtFex(1
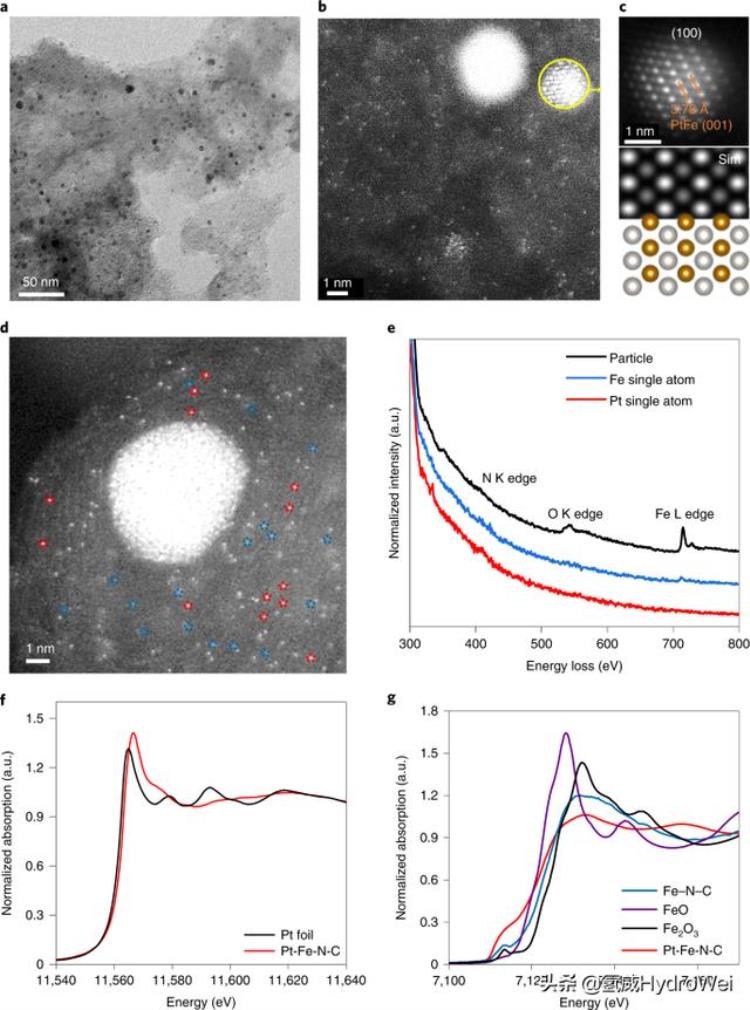
图1:Pt-Fe-N-C电催化剂中多个活性位点的表征。
检测到两组明亮的孤立斑点,通过其对比度来区分(图1d和补充图6),并且可以归因于Pt和Fe单个原子,因为它们在HAADF-STEM中的Z对比度差异17.通过局部电子能量损失光谱(EELS)分析进一步证实了这些不同分离点的鉴定(图1e和补充图6)。纳米粒子的EELS图谱显示出很强的Fe和O信号,由于Pt主峰的高能量损失(约2,200 eV)而无法检测到Pt信号。18.蓝色虚线圆圈中单个原子的轮廓显示出弱的Fe和N信号,这表明较弱的点是Fe-N部分。相比之下,红色虚线圆圈中单个原子的轮廓仅显示弱N信号,这表明较亮的点是Pt-N部分。这些结果与STEM图像中的对比度差异非常一致。微观表征清楚地表明了杂化材料中丰富的Pt和Fe单原子以及Pt-Fe纳米颗粒的共存。
根据电感耦合等离子体质谱结果,Pt-Fe-N-C中的金属负载为2.0 wt%铁和 1.7 重量%铂.在X射线光电子能谱表征中,Pt 4f光谱的反卷积为Pt的主峰0和 Pt2 以及少量的铂4 (附图7a)。将Fe 2p谱解析为Fe的双峰2 和Fe的三重峰3 (补充图7b)。同时,反卷积的N 1s谱(补充图7c)包括一个显性石墨N以及吡啶、金属和氧化N。与Fe-N-C相比,Pt-Fe-N-C催化剂中吡啶N的强度降低(附图8)是由于前者的两步高温热解。采用X射线吸收光谱(XAS)进一步表征了Pt-Fe-N-C和Fe-N-C的结构(补充图9)。图1f,g比较铂L3-边缘和铁K边缘,分别是Pt-Fe-N-C的X射线吸收近边缘结构光谱,具有各种标准。Pt L没有明显差异3边缘(图1f)在前边缘区域与金属Pt箔相比。Pt-Fe-N-C的白线强度更强,这是由Pt 5d到Fe 3d轨道的电子转移引起的,表明Pt在Pt-Fe-N-C中处于氧化形式( ref.19).在扩展的X射线吸收精细结构数据的傅里叶变换中,主峰从Pt箔的2.42 Å到Pt-Fe-N-C的2.23 Å的负偏移(补充图10a)明确肯定了Pt-Fe键的形成,而1.5 Å处的弱峰可能是由Pt-N构型贡献的。对于图1g中的Fe K边缘,Pt-Fe-N-C样品的前边缘峰强度最高,而白线强度与Fe-N-C和Fe氧化物相比显着降低。与Fe-N-C相比,Pt-Fe-N-C中Fe的前边缘特性的损失(补充图9)表明前者中的大多数Fe金属中心失去了八面体对称性20.2.06 Å处的强傅里叶变换峰是由于Pt-Fe合金的散射,而1.50 Å的肩部是由碳载体中Fe-C/N/O键的散射引起的(补充图10b)。Pt和Fe元素的配位信息得到了相对拟合结果的进一步支持(补充图11和补充表1)。
57应用Fe Mössbauer光谱法鉴定了Fe物种的局部结构。如补充图12和补充表2所示,Fe–N–C的莫斯堡尔谱拟合了两个主控双态和少量分配给γ-Fe的单线态。双精度体 D1 和 D2 被分配到方形平面 Fe(II)N4分别与Fe(II)在低自旋和中自旋状态下协调21,22.在Pt添加和两步热处理后,双层和单层的面积分别减小和增加。57Fe Mössbauer对Fe合金的研究,如Pt-Fe(参考文献23),Ru–Fe(参考。24)和Pd–Fe(参考文献。25,26)表明Fe在合金中是零价的。巴塞洛缪和布达特23结果表明, 合金中表面Fe原子的默斯堡尔吸收概率与本体中的吸收概率基本相同, 特别是对于纳米级的Pt-Fe颗粒.因此,Pt-Fe-N-C中的单线态成分主要是由于Pt-Fe合金,而D1和D2双态态来自氮配位的Fe单原子。
铂铁-N-C的性能评估Pt-Fe-N-C在Ar饱和0.1 M HClO中的循环伏安曲线4由于Pt负载超低,电解质没有表现出深的氢吸附和/或解吸峰(补充图13)。Pt-Fe-N-C催化剂的ORR性能通过O形旋转环盘电极进行评价2-饱和 0.1 M 盐酸4铂含量为 4.25 μg cm 的电解质–2,不带背景或 iR 校正。补充图14显示,Pt-Fe-N-C的ORR活性比Fe-N-C显着提高,因为半波电位从0.765 V转移到0.909 V。Pt-Fe-N-C在0.9 V时的相应Pt MA达到1.74 A mg铂–1,比Pt / C(0.18 A mg)高约十倍铂–1).此外,在较宽的电位窗口中,Pt-Fe-N-C的环电流远低于Fe-N-C的环电流(补充图14)。计算出的最大过氧化氢产率(H2O2%)和电子转移数分别为2.4%和4,这表明发生了完整的四电子转移反应。
在 O 中评估了 Pt-Fe-N-C 催化剂的耐久性2-饱和 0.1 M 盐酸4电位循环期间的电解质范围为 0.6 至 1.0 V。Pt-Fe-N-C的半波电位(补充图15)在0.6至1.0 V之间循环40,000次后仅下降14 mV,远优于Fe–N–C(16 mV)(参考文献)。27) 和 Pt/C (10 mV) 仅经过 10,000 次循环(补充图 16)。未观察到颗粒聚集,根据测试后拍摄的HAADF-STEM图像,Fe和Pt单个原子的均匀分布得到很好的保留(补充图17和18)。能量色散X射线光谱(EDX)映射和相应的元素线(补充图18)进一步证实了Pt-Fe纳米颗粒在40,000次循环后保持高度稳定的有序结构和Pt壳的形成。
在 H 中评估了 Fe–N–C、Pt–N–C、Pt/C 和 Pt-Fe-N-C 催化剂的燃料电池性能2/O2环境,并在图2a中进行比较。截止电流密度为 2 A cm–2用于所有测试。阳极的Pt负载为0.1 mg cm–2在所有测量中;阴极的Pt负载量为0.015 mg cm–2对于Pt-Fe-N-C,和0.1毫克cm–2对于 Pt–N–C 和 Pt/C。Pt–N–C和Fe–N–C的燃料电池达到0.32和0.66 W cm的峰值功率密度–2,分别与文献中报告的值相当28,29.燃料电池在阴极中Pt-Fe-N-C的性能显着增强,因为它达到了1.08 W cm的功率密度–2在 2.0 A 厘米–2.即使功率密度低于Pt / C(1.37 W cm–2),值得注意的是,后者的Pt负载高出七倍。H 中的 Pt-Fe-N-C 电池2/空气环境(补充图19)在整个电流密度范围内表现出比Fe-N-C更好的性能,并且峰值功率密度更高(0.55对0.33 W cm–2).Pt-Fe-N-C的Pt质量活性校准为绝对H2和 O2压力为 1 bar(详细信息如方法所示)从 H2/O2和 H2/空气极化曲线分别为0.77和0.74 A mg铂–10.9 V 时无 iR,分别比Pt / C(0.21 A mg)高出约3.7倍铂–1),比2025年活动目标(0.44 A mg)高出1.75倍铂–1)由美国能源部(DOE)设定。Pt-Fe-N-C的MA高于大多数Pt基(补充表3)和杂化电催化剂(补充表4),仅略低于Pt-Co/Co-N-C(1.07 A mg)铂–1,校准为绝对 H2和 O2压力为1巴)由Chong等人报告。11.虽然Pt-Fe-N-C催化剂实现了较高的MA,但由于Pt负载量低得多,它仍然无法在高电流密度区域与Pt基电催化剂竞争。
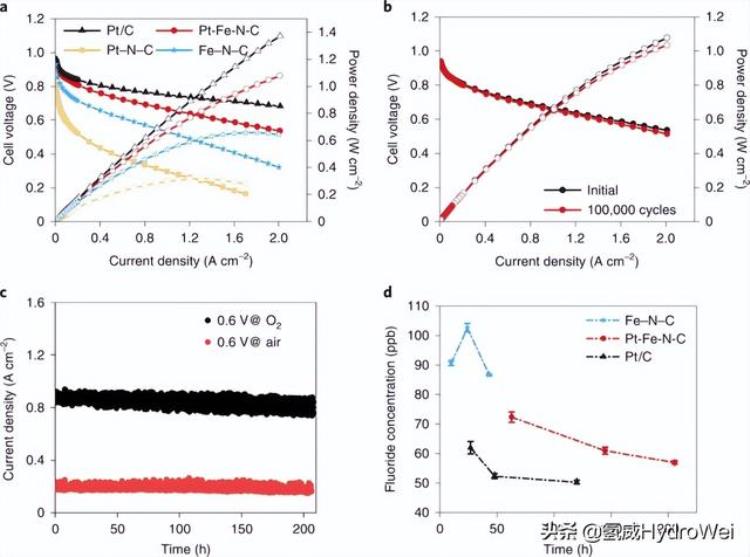
图2:燃料电池中Pt-Fe-N-C阴极的性能评估。
按照DOE测试方案,将Pt-Fe-N-C,Pt / C和Fe-N-C阴极组装的电池在0.6和0.95 V的重复方波循环下进一步进行加速耐久性测试,在每个电位下保持3 s。如图2b所示,Pt-Fe-N-C阴极的燃料电池极化曲线在100,000次循环后显示出可忽略不计的变化。0.9 V 时的 MA无 iR和 2.0 A cm 时的功率密度–2略微降低至0.75 A mg铂–1和 1.03 瓦厘米–2即使在100,000次循环之后,分别对应于3%和5%的下降。这些结果超过了DOE的耐久性目标,即在30,000次循环后MA损耗小于40%。相比之下,Pt/C(补充图20a)和Fe–N-C(补充图20b)阴极在30,000次循环后显示出显着的性能损失。Pt/C的Pt MA从0.21 A下降到0.10 A mg铂–1Fe–N–C的峰值功率密度从0.66 W显著降低到0.56 W cm–2.
采用 STEM-EELS 进一步分析了Pt-Fe-N-C催化剂在燃料电池中循环100,000后的形貌和结构.丰富的单个原子仍然均匀地分布在碳载体上(图3a,b)。EELS分析(图3b)验证了具有N配位构型的Pt和Fe单个原子的保存,这与原始样品相似(图1d)。没有观察到Pt-Fe纳米颗粒的明显聚集(补充图21)。纳米颗粒的结构和组成在潜在的循环过程中确实发生了变化。总的来说,Pt-Fe纳米颗粒的结构演化遵循两条途径。当粒径小于~4 nm时, 形成固体PtFe@Pt核壳结构(图3c), 晶格间距为0.191 nm,核心PtFe(002)为0.204 nm,壳层中Pt(002)为0.204 nm, 源自原始的PtFe阶结构.线强度曲线进一步支持了这一结论(图3d)。观察到中心强度的周期性振荡和壳的单调性,这可以归因于Pt和Fe在有序晶格中的对比度差异。在最外层的三层,强度振荡消失,晶格膨胀,这证实了Pt壳的形成。但是,PtFe越大x(1
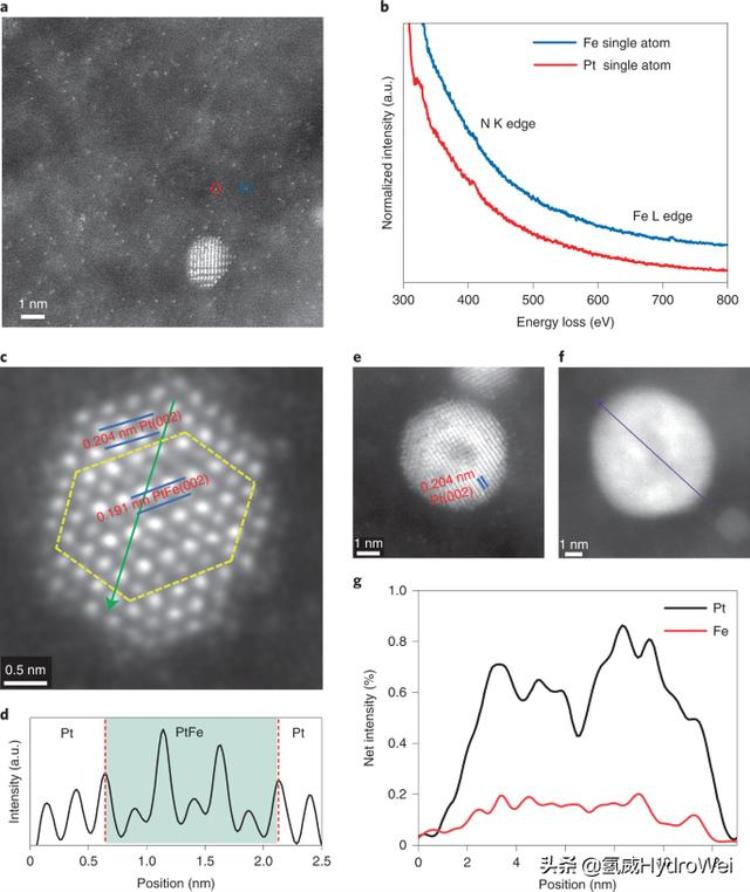
图3:耐久性测量后Pt-Fe-N-C电催化剂的表征。
在0.6 V电压下,在1 bar绝对压力下对阳极和阴极进行了计时安培测试,以进一步评估不同阴极催化剂的长期耐久性。如图2c(红线)所示,燃料电池具有Pt-Fe-N-C阴极(Pt负载量为0.015mg铂厘米–2)在H中显示出206小时内几乎恒定的电流密度2/空气环境。相比之下,Pt / C电池的电流密度(Pt负载为0.1mg铂厘米–2) 在 120 小时内下降了 8%(补充图 24a)。Pt-Fe-N-C 在 0.9 V 时的 Pt MA无 iR归一化至 1 bar 绝对压力从 0.72 A 略微增加到 0.75 A mg铂–1.这种增强可能是由于Pt-Fe纳米颗粒的结构演变(到更理想的核壳结构)。对于Fe–N–C(补充图24b),电流密度下降更快,43小时后损耗为73%,与文献中报告的结果相似。33,34.在H中也进行了类似的测试2/O2环境。在0.6 V下电位保持210小时后,Pt-Fe-N-C电池的电流密度仅下降了5%(图2c,黑线)。轻微的电流密度下降可能是由于水淹。0.85 V时的电流密度在两个H中也显示出类似的小幅下降。2/空气和 H2/O2测试(补充图25)。电位循环和恒压试验均证实,Pt-Fe-N-C比Pt/C、非Pt族金属、Pt基(补充表3)和其他杂化电催化剂具有更好的燃料电池耐久性(补充表4)。
铁是Nafion基膜和离聚体的关注点,因为芬顿反应35.The F−监测出水中0.6 V的浓度作为时间的函数,以评估其降解速率。如图2d所示,Fe–N–C总是具有较高的F−浓度比Pt/C和Pt-Fe-N-C催化剂高.F的浓度−在Pt-Fe-N-C中略高于Pt / C,可能是由于前者中更多的离子体。Pt-Fe-N-C浓度持续下降,达到接近200 h以上Pt/C的水平。这一结果清楚地表明,Pt-Fe-N-C细胞中膜和离聚体的降解速率远慢于Fe-N-C细胞中的膜和离聚体,部分原因是H水平较低。2O2在前者中。
理论研究采用密度泛函理论(DFT)计算, 探索了混合电催化剂高活性和耐久性的起源.构建了几个模拟模型来表示活性位点的可能类型(补充图26),其中包括单个原子(Pt-N1C3, 铂–N2C2和 Fe–N1C3根据扩展的X射线吸收精细结构拟合结果补充表1),Fe-Pt双金属和Pt毫升/PtFe(111) 核壳结构(PtFe(111) 基板上的单层 (ML) Pt 皮肤)。Pure Pt(111) 也被包括在内进行比较。在这些模型中,Pt–N2C2在结构优化和能量计算的基础上,对Fe-Pt双金属进行了精心设计和选择(补充图27和28以及补充表5)。为了评估各种仿真模型的ORR途径,优化了关键中间体*OOH,*O和*OH(*表示吸附状态)。反应中间体在碳基模型(Pt–N)上的相应吸附结构1C3, 铂–N2C2, 铁–N1C3和Fe-Pt双金属)如图29所示,而Pt上则显示了毫升/PtFe(111) 如补充图 30 所示。4e的ORR的吉布斯自由能图−在U处构建了各种可能活性位点的通路断续器= 0.9 V(RHE,可逆氢电极),对应于实验中活性评估的电位。如图4a和补充表6所示,Pt–N中的单个Pt金属位点1C3(绿色曲线)表现出最佳的ORR活性,除最后的*OH质子化步骤外,所有基本反应均呈下坡趋势,ORR期间的能量势垒仅为0.17 eV,远小于Fe-N中单个Fe位点的能量势垒1C3(0.53 eV,图4a中的黑色曲线)。对于另外两个碳基模型,Pt–N2C2作为电位限制步骤(红色曲线),*OOH形成(0.54 eV)显示出非常高的能垒,Fe-Pt双金属(蓝色曲线)也是如此,由于非常强的OH结合能使得*OH去除非常困难(0.75 eV),预计不会非常活跃。铂毫升/PtFe(111)也被预测为对ORR非常活跃,因为在最后的*OH质子化步骤中具有0.18 eV的低能垒(图4a中的紫色曲线),这与Pt-N的能垒接近。1C3(0.17 eV)。请注意,这里的能量势垒只是热力学能量势垒,除Pt-N上的解吸外,*OH的解吸是吸热的2C2.这种具有1 ML Pt皮肤的核壳结构显示出比Pt(111)更好的ORR活性(图4中的黄色曲线),因为与反应中间体的结合较弱,特别是对于*O和*OH。在潜在循环后,PtFe@Pt纳米颗粒中Pt壳层的厚度可以从一到三个原子层不等。因此,在2Pt上也进行了模拟。毫升/PtFe(111) 和 3Pt毫升/PtFe(111).如补充图31所示,所有Pt皮肤都显示出比纯Pt更好的ORR活性。其中,Pt毫升/PtFe(111) 是最好的,*OH-to-H 的势垒最低2O步骤。根据我们的计算,Pt–N1C3在表面和地下浸出Fe后形成的核壳纳米颗粒被认为是Pt-Fe-N-C中最具活性的位点。
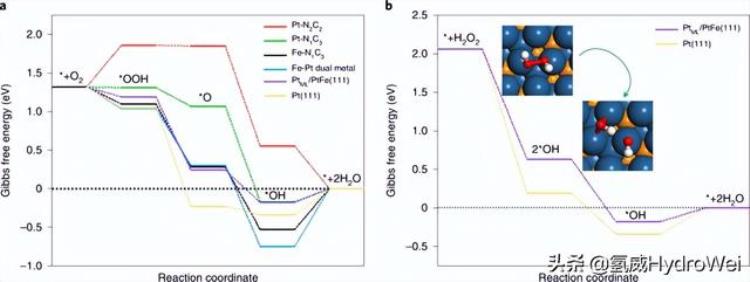
图4:Pt-Fe-N-C电催化剂性能增强的理论研究.
燃料电池中Me-N-C催化剂的主要问题之一是形成大量的H2O2作为最终产物,对膜和离聚体有害36.发现混合Pt(引用。12,37),铂钴合金11和/或CeOx(参考资料)38)颗粒作为Me-N-C的过氧化物/自由基清除剂可以减轻H2O2积累。因此,在我们的杂化催化剂中,H2O2在Fe–N–C(补充图32)甚至Pt-N-C位点(补充图33)上产生的也可以进一步减少到H2O在附近的Pt-Fe纳米颗粒上。为了验证这一假设,H2O2减少对铂毫升比较了/PtFe(111)和Pt(111)表面(图4b和补充表7)。原来Pt毫升/PtFe(111) 为快速 H 做出了贡献2O2-到 2*OH 转换和最终的 *OH-到 H2O阶跃也很简单,能量势垒为0.18 eV,低于Pt(111)(0.34 eV)。。因此,我们期望所有三个活性位点,即Pt-N1C3, 铁–N1C3PtFe@Pt,有助于在Pt-Fe-N-C中实现ORR的高性能。在燃料电池运行期间,在PtFe@Pt上形成耐用的Pt壳,并进一步降低H2O2单原子活性位点产生的PtFe@Pt是其超强耐久性的主要原因。
结论综上所述, 成功合成了一种由原子分散的Pt和Fe单原子以及Pt-Fe合金纳米颗粒组成的Pt负载量超低(1.7 wt%)的杂化ORR电催化剂.良好的性能,包括0.77 A mg铂–1在绝对 H 时2和 O20.9 V 时压力为 1 bar无 iR和 1.08 W 厘米–22.0 A cm 时的功率密度–2,是在燃料电池中实现的。更重要的是,这种混合电催化剂表现出优异的耐久性,在0.6和0.95 V下循环100,000次后具有97%的活性保留,并且在200小时内0.6 V时没有明显的电流下降。理论模拟表明,Pt–N1C3, 铁–N1C3PtFe@Pt都是ORR的活跃部位。混合电催化剂的耐久性增强可能是由于H降低2O2膜的形成和随之而来的缓解和离聚体的降解。我们的研究结果强调了混合电催化剂中不同活性位点之间协同效应的重要性,并为为燃料电池和其他电化学器件设计更活性和更耐用的低Pt基团金属电催化剂提供了另一种方法。
方法化学品和试剂硝酸锌六水合物(锌(NO3)2·6H2O, 阿拉丁, 99.998% 金属基), 七水合硫酸亚铁 (FeSO4·7H2O, 阿拉丁, 99.95% 金属基), 2-甲基咪唑 (C4H6N2, 阿拉丁, 98%), 1,10-菲咯啉一水合物(C12H8N2, 阿拉丁, 99%), 铁氰化钾 (K3铁(中国)6,阿拉丁,99.95%金属基),氢氧化钾(KOH,阿拉丁,99.99%金属基),乙酰丙酮铂(II)(Pt(C)5H7O2)2,西格玛奥德里奇,≥99.98%痕量金属基础),含有异丙醇(C3H8O,Fisher,99.8%)和Nafion 117(Sigma-Aldrich,在低级脂肪醇和水的混合物中为5重量%),甲醇(CH3OH,Scharlau,99.8%)和无水乙醇(C2H5OH, VWR, 99.5%), 高氯酸4,GFS化学品,70%的veritas双蒸馏)被购买并直接使用,无需进一步纯化。D521 Nafion分散体(EW1100,基于酒精),气体扩散层(GDL,科德宝H14C7,由25μm微孔层组成)和Nafion HP膜(CL-2019-50,20μm)是从燃料电池商店(https://www.fuelcellstore.com)购买的。商用Pt/C催化剂(46.4重量%,TEC10E50E)来自TKK。
催化剂合成铁掺杂ZIF-8的合成细节可以在别处找到27,39.简而言之,0.95 mM 锌(NO3)2·6H2O 和 0.05 mM 铁氧烷4·7H2O作为金属源,以8.21克2-甲基咪唑为有机连接剂,与甲醇混合形成Fe掺杂的ZIF-8。通过用无水乙醇洗涤产物三次来除去多余的有机连接物和金属源。在80°C真空烘箱中干燥过夜后,将Fe掺杂的ZIF-8前驱体直接在1,000°C的Ar气体(纯度99.99%)中热处理1 h,得到最终的Fe-N-C催化剂。原子分散的Fe原子均匀分布在N-C框架上,无需进一步的酸洗。最终催化剂中Fe负载的重量约为2.0 wt%。
根据我们之前的工作,用超低Pt负载浸渍Fe-N-C可以显着提高Fe-N-C的耐久性,但它并没有表现出明显的活性改善,特别是在动力学区域39.在这项后续工作中,氨热处理旨在暴露更多的活性位点,并定制碳载体的性质和单个原子的配位数。氨处理后的二次Ar热解旨在稳定碳框架。因此,在这项工作中,铁掺杂的ZIF-8被用作Pt浸渍的支持,以简化热处理方案。通过超声将乙酰丙酮(10mg)Pt(II)均匀分散在3ml乙醇中,直到溶剂变得透明。同时,将110mg溶解在10ml乙醇中的1,10-菲罗啉一水合物加入到Pt溶液中,为Pt配位提供足够的氮源。将约400mg的Fe掺杂的ZIF-8加入到上述溶液中以形成均匀的悬浮液。在真空烘箱中以60°C干燥过夜后,收集固体并球磨(ZrO2球,每分钟350转(r.p.m.),4小时),以均匀分布铁掺杂的ZIF-8支架上的Pt和N源。混合前体首先在NH中处理3气体在900°C下放置15分钟,然后在1,000°C的Ar气氛中转移1小时,以除去前驱体中的Zn并稳定整个碳框架以产生最终催化剂Pt-Fe-N-C。铂和铁的负载量分别约为1.7%和2.0重量%。
作为参比电催化剂, 由 ZIF-8 载体制备了 Pt–N–C, 该载体由 1 mM Zn(NO3)2·6H2O和8.21克2-甲基咪唑在甲醇溶剂中,并遵循与Fe掺杂的ZIF-8相同的收集和干燥方案。类似地,通过超声将10mg乙酰丙酮铂(II)均匀分散在3ml乙醇中,直到溶剂变得透明。同时,将110mg溶解在10ml乙醇中的1,10-菲罗啉一水合物加入到Pt溶液中,为Pt配位提供足够的氮源。向该溶液中加入约400mg ZIF-8以形成均匀的悬浮液。在真空烘箱中以60°C干燥过夜后,收集固体并球磨(ZrO2球,350 r.p.m.,4 小时)以均匀分布 ZIF-8 支架上的 Pt 和 N 源。混合前体最初也在NH中处理3气体在900°C下15分钟,然后在1,000°C下转移到Ar气氛中1小时。最终的催化剂Pt–N–C的Pt负载量为2.3 wt%。
物理表征使用配备Gatan Enfina EELS的300 kV下工作的双Cs校正FEI Themis G2收集TEM结果,HAADF探测器的收集角为60-200 mrad,探针电流控制在150 pA左右。EELS的收敛半角和集合半角分别为25和36.2 mrad。EELS的采集时间限制在每像素0.1 s,因此使用Fischione 2550冷冻转移断层扫描支架来最小化单个原子的跳跃。Pt-Fe-N-C的结构由配备石墨单色仪和Cu Kα辐射源的X射线衍射仪(PANalytical,X'pert Pro)进行检查。分别通过电感耦合等离子体质谱(安捷伦技术公司,安捷伦7900)和X射线光电子能谱(Kratos Analytical,Axis Ultra DLD)评估体积和表面组成。催化剂的表面积通过Brunuer-Emmett-Teller方法(Quantachrome Instruments,AUTOSORB-1)测量。采用Si(111)单色仪在阿贡国家实验室先进光子源光束线20-BM上进行了XAS表征,包括X射线吸收近边缘结构光谱和扩展X射线吸收精细结构光谱。XAS数据使用雅典娜和ARTEMIS软件包进行处理。这57Fe–N–C 和 Pt-Fe-N-C 的 Fe Mössbauer 光谱在 SEE Co W304 Mössbauer 光谱仪上记录,使用57透射几何结构中的 Co/Rh 源。数据是使用MOSSWINN 4.0软件安装的。微拉曼光谱(雷尼绍,InVia)用于探测耐久性测试前后碳载体的变化。通过离子色谱法(瑞士万通881与紫外和电导率检测器)分析从阴极气体出口收集的出水样品,以评估氟化物的排放。
电化学测量为了制备催化剂油墨,将2.5mg催化剂均匀分散在500μl混合溶剂(水和异丙醇以4:1体积比)和10μl5重量%Nafion 117溶液中。用铝抛光旋转环圆盘电极(直径5.5 mm)2O3粉末(50纳米)。将一定量的催化剂墨水滴到电极上。在空气中干燥后,通过电化学工作站(CHI 760E)评估薄膜电极。碳棒和Ag/AgCl分别用作计数器电极和参比电极。所有电位都参考了 RHE。在每次活性测试之前,都校准了参考RHE的Ag / AgCl电极的电位。在校准过程中,将Ag / AgCl电极和Pt箔放入H2-饱和 0.1 M 盐酸4然后记录电解质,以及这两个电极之间的电压差。以这种方式测量的Ag / AgCl值约为0.26-0.27 V断续器.
循环伏安图(20个周期),电位范围为0–1.2 V,100 mV s–1应用于在Ar饱和的0.1M HClO中清洁薄膜4解,然后在50 mV s的相同电位范围内取稳定的循环伏安图曲线–1.ORR 性能以 O 为单位进行测量2-饱和 0.1 M 盐酸4在 5 mV s 下旋转速率为 1,600 r.p.m. 的溶液–1.在活动评估过程中采用线性扫描伏安技术,记录的偏振曲线均为0.125~1.0 V。在耐久性测试中,Pt-Fe-N-C 和 Pt/C 在 50 mV s 下承受了 0.6 至 1.0 V 之间的潜在循环–1在 O 中2-饱和 0.1 M 盐酸4电解质。
旋转环圆盘电极的收集效率首先在Ar饱和的10 mM K中测定3铁(中国)6 1 M KOH 溶液。电极以1,600 r.p.m.旋转(对应于测量过程中施加的角速度),并通过分别将环形和圆盘电压设置为1.5和0.1 V来进行电化学计量i-t测量。

原文:科大研发全球最耐久的氢燃料电池 | 香港科技大学 (hkust.edu.hk)
Atomically dispersed Pt and Fe sites and Pt–Fe nanoparticles for durable proton exchange membrane fuel cells | Nature Catalysis
文章评论